Difference Between Amyloid and Prion
Amyloids and prions are both synonymous with neurodegenerative disorders, disorders that cause degeneration within the brain. Amyloids are not brain specific, as they can cause disorders in any organs in the body. In terms of neurology, the human neurodegenerative disorders for which amyloids and prions are directly or indirectly responsible, are those such as Alzheimer’s disease, Parkinson’s disease, and Creutzfeldt-Jakob disease. Understanding the workings within amyloids forms an important aspect of understanding prions and neurodegeneration.
Difference between Amyloid and Prion
Definition and Pathophysiology
Amyloids are aggregates (a formation made from many parts compiled together) of a rod-like structure which contains repetitions of a certain type of protein. When abnormal plasma cells stemming from the bone marrow form abnormal light chain proteins, they enter the bloodstream and form amyloid deposits. These deposits can cause a build up in any vital organ in the body (such as the brain, heart or intestines), resulting in various serious medical conditions.
Prions are an abnormal form or folding of the specific proteins of amyloids deposited in the brain, making them infectious and able to renew indefinitely. In other words, prions are defined as a subclass of amyloids where protein aggregation has come infectious and changed the state of self-production. The prions have the ability to transmit their misfolded protein shape into the same proteins occurring in a normal state.
Prions are responsible for the neurodegenerative disorders that can occur in humans and animals. This is due to the misshaped protein being more difficult to break down by enzymes and end up accumulating in the neurons instead, resulting in destruction. When this neuron destruction progresses, it gradually causes tissue of the brain to have a sponge-like pattern filled with holes.
Potential causes
Amyloids occur when bone marrow produces abnormal blood plasma cells. These abnormal plasma cells then form abnormal types of light chain proteins. When the abnormal light chain proteins enter the circulation system (blood stream), they deposit into vital organs throughout the body. This can be due to:
- Genetic or hereditary components (which affect the eyes, heart, kidneys and the brain)
- Chronic diseases and types of cancers
- Cell mutations
- Rare infections
Prions are the misshaped proteins causing amyloids deposited in the brain. Genetically, the PRNP gene is responsible for directing the body to product prion protein. When this gene mutates it can cause abnormal protein production, resulting in specific prion diseases of the brain. Prion disease can also occur sporadically, resulting in other specific diseases of the brain.
Symptoms
Symptoms of abnormal amyloid protein deposits vary and depend on which tissue or organ has been affected by the deposits.
Prion disease, caused by misshaped prion proteins resulting in amyloids in the brain, often presents the following symptoms:
- Memory difficulties and changes in judgment
- Personality changes
- Confusion, difficulty with speech, and/or disorientation
- Changes in coordination and involuntary muscle spasms
- Decreased quality of vision or blindness
Diagnosis
Diagnosing the occurrence of amyloids in the affected organs specifically can be done through biomarker tests. These include measuring the changes in the size of the organ and its functions, measuring levels of specific proteins in the blood, in cerebrospinal fluid, and on scans (MRI, CT, PET).
Diagnosing prion proteins in the brain causing prion disease can be done through scans of the brain to measure function and size, cerebrospinal fluid testing for specific prion disease markers and neurodegeneration markers, and EEG to record electrical activity changes in the brain.
Treatment
Treating amyloids depends on where they occur. The only way to completely abolish amyloids from being produced is to undergo intensive chemotherapy to destroy abnormal blood cells in the bone marrow responsible for amyloid production. There is no other way to treat amyloids and the disorders caused by the occurrence of them are treated on a specific basis.
Prions causing prion disease can be treated with medications, assisted living associated with neurodegeneration, upholding hydration and nutrient intake. This does not cure the production of prions or neurodegeneration but provides a way for improving quality of life and treating associated symptoms.
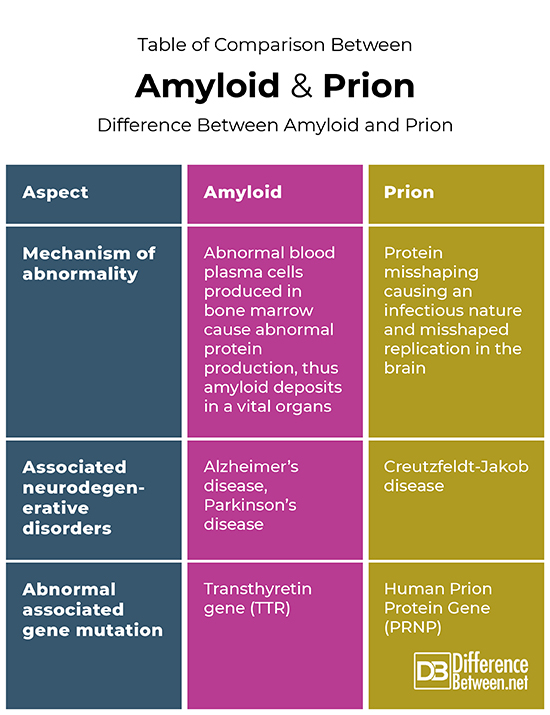
Table of comparison between amyloid and prion
Summary of Amyloid vs Prion
Amyloids and prions are both associated with protein misshaping and certain neurodegenerative disorders. In the case of amyloids, misshaped proteins can cause disorders in any organ where they are deposited. Prions, however, are the misshaped proteins that can cause amyloids in the brain and have associated disorders limited to the brain. In terms of neurodegenerative disorders, amyloids are responsible for Alzheimer’s disease and Parkinson’s disease, where prions are responsible for Creutzfeldt-Jakob disease.
FAQ
Are prions a type of amyloid?
Amyloids are aggregates with a fibrillar structure that is constituted by repetitions of a specific protein. Prions are defined as a sub class of amyloids. With prions, the protein aggregation becomes self-perpetuating and then becomes infectious – this is the cause of many fatal neurodegenerative diseases such as Creutzfeldt-Jacob disease.
How are amyloid plaques related to prions?
Amyloid plaques have been found in the brains of people with Creutzfeldt-Jacob disease. Creutzfeldt-Jacob disease is an illness caused by prions. Scrapie is another prion disease, this time found in animals, with amyloid plaques that are composed of prion proteins
What is the difference between a prion and a protein?
The main difference between a protein and a prion is in the structure – prions are misfolded causing them to be able to change the shape of other similar proteins, thus becoming infectious.
Is amyloid beta a prion?
Due to its ability to self-propagate as well as the existence of several distinct ‘strains’, amyloid beta shares a lot of indistinguishable properties with prions. These properties are what is likely making it possible to become a prion during diseases.
- Difference Between a Cochlear Implant and Normal Hearing - October 4, 2022
- Difference Between Obstructive and Restrictive Spirometry - September 11, 2022
- The Difference Between White Box and Black Box Testing - September 11, 2022
Search DifferenceBetween.net :
Leave a Response
References :
[0]Kitamoto, T., Tateishi, J., Tashima, T., Takeshita, I., Barry, R.A., DeArmond, S.J and Prusiner, S.B. Amyloid plaques in Creutzfeldt-Jakob disease stain with prion protein antibodies. Annals of Neurology. 1986, vol. 20, no. 2, pp: 204-208.
[1]Sabate, R., Rousseau, F., Schymkowitz, J., Batlle, C and Ventura, S. Amyloids or prions? That is the question. Prion. 2015, vol. 9, no. 3, pp:200-206.
[2]Watts, J.C and Prusiner, S.B. β-Amyloid Prions and the Pathobiology of Alzheimer's Disease. Cold Spring Harb Perspect Med. 2018, vol.8, no. 5, pp: a023507